Research
Overview
The Griffith lab is a combined research group driven by the interests of twin scientists Malachi Griffith and Obi Griffith. The focus of the lab is on developing methods of applied bioinformatics for personalized medicine and improved cancer care.
Our research is committed to developing open-access and open-source resources for cancer genome analysis. Research projects cover a wide spectrum of cancer informatics and clinical statistics with an emphasis on translation and application. Specifically, we use computational methods for the analysis of large cancer datasets at the molecular level (DNA, RNA, and protein) to identify markers for diagnosis, prognosis, and drug response prediction in cancer. We have contributed to the early development of methods for the analysis of transcriptional regulation (ORegAnno) and RNA-seq analysis and visualization (ALEXA-seq Platform).
The group is engaged in a large number of tumor sequencing projects for breast, lymphoma, glioblastoma, lung, and other cancers, investigating primary, relapse, and drug-resistant tumors. To this end, we have worked with others at the McDonnell Genome Institute to develop end-to-end pipelines for clinical cancer sequencing that automate state-of-the-art methods for sequence alignment, somatic variation detection, RNA sequence analysis, and the integration of these data types into user-friendly reports of the most clinically relevant genome and transcriptome changes in a tumor or cohort of tumors (Genome Modeling System). To aid in this effort our group has developed software, databases, knowledgebases, and web tools for interrogation of the druggable genome (DGIdb), identification of cancer driver mutations (DoCM), interpretation of clinically actionable variants in cancer (CIViC), and genomic visualization (GenVisR). The group is also actively involved in the identification and scoring of tumor neoantigens and the development of related software for the design of human cancer vaccines (pVACtools). We are also working with organizations such as the Global Alliance for Genomics Health (GA4GH) to help define data models and standards for clinical translation of genomics data, in particular through our leadership of the driver project: Variant Interpretation for Cancer Consortium.
In addition to our basic and clinical research interests, we are also passionate about the scholarship of teaching and learning. We have made substantial contributions to the training and education of tomorrow’s bioinformaticians through our involvement in CBW, CSHL workshops and the BioStars forum. We have developed online courses for RNA sequence analysis, Genomic Visualization in R, and precision/personalized medicine bioinformatics.
Breast cancer genomics
Genomic analysis of the Stat1-/- mouse model of ER+ breast cancer
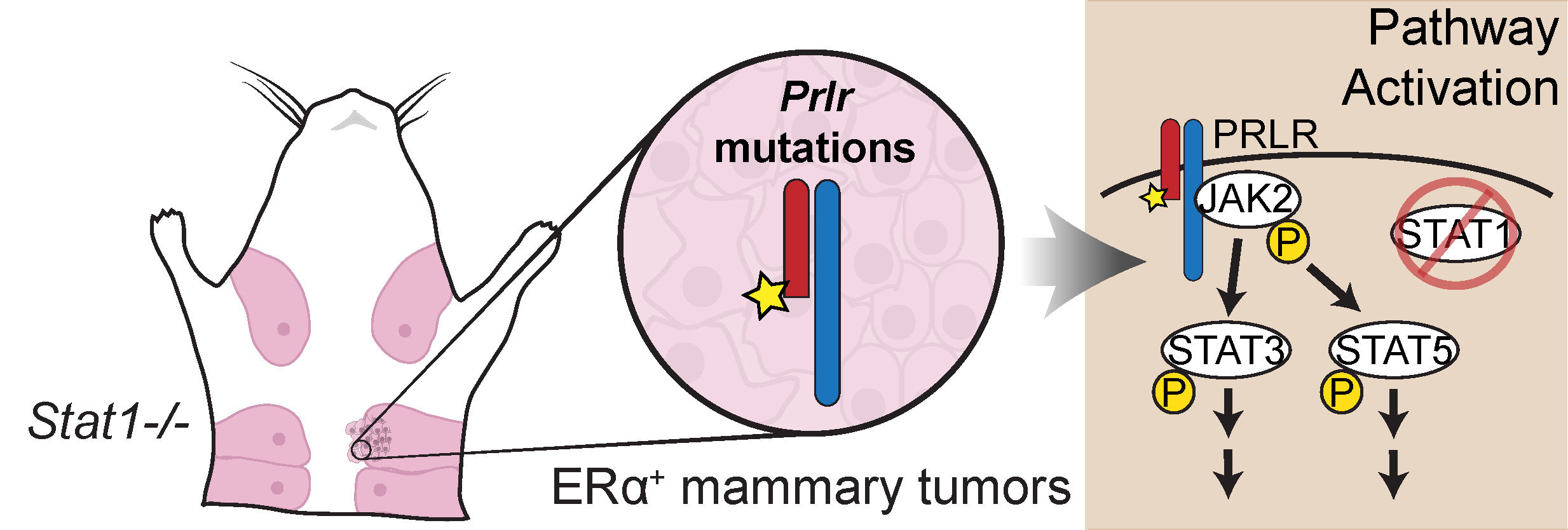
Description:
ER+ luminal tumors are the most frequent subtype of breast cancer. Stat1−/− mice develop mammary tumors that closely resemble this cancer subtype. To identify transforming events that contribute to tumorigenesis, we performed whole genome sequencing of Stat1−/− primary mammary tumors and matched normal tissues. This investigation identified somatic truncating mutations affecting the prolactin receptor (Prlr) in all tumor samples. Targeted sequencing confirmed the presence of these mutations in precancerous lesions, indicating this is an early event in tumorigenesis. Functional evaluation showed that co-expression of truncated and wild-type Prlr led to aberrant Stat3 and Stat5 activation downstream of the receptor, cellular transformation in vitro and tumor formation in vivo.
Publication(s):
Team members:
Obi Griffith, Malachi Griffith, Kilannin Krysiak, Zachary Skidmore, Jasreet Hundal, Lee Trani
Liver cancer genomics
A genomic case study of mixed fibrolamellar hepatocellular carcinoma
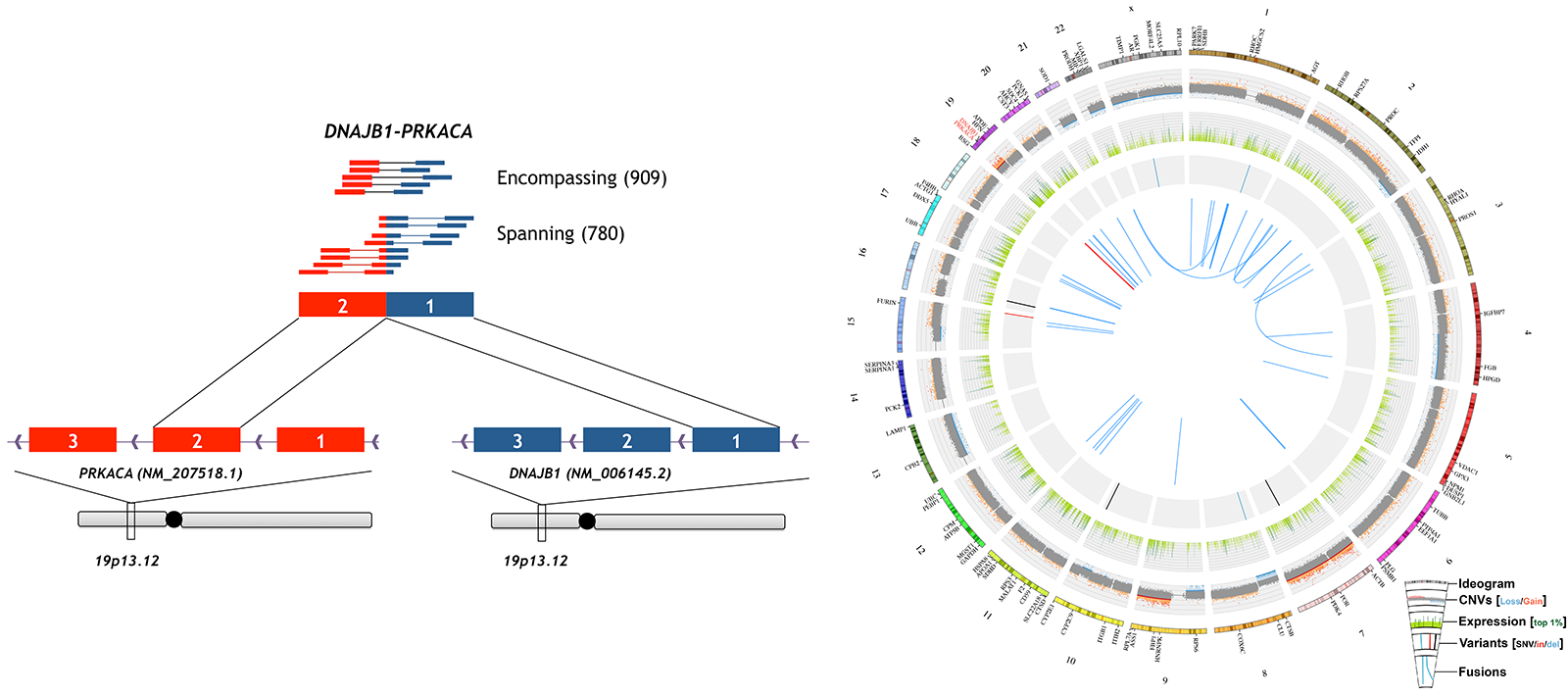
Description:
We reported the first comprehensive genomic analysis of a case of Mixed fibrolamellar hepatocellular carcinoma (mFL-HCC). No common HCC-associated mutations were identified. The very low mutation rate of this case, large number of mostly single-copy, long-range copy number variants, and high expression of ERBB2 were more consistent with previous reports of pure FL-HCC than conventional HCC. In particular, the DNAJB1:PRKACA fusion transcript specifically associated with pure FL-HCC was detected at very high expression levels. Subsequent analysis revealed the presence of this fusion in all primary and metastatic samples, including those with mixed or conventional HCC pathology. A second case of mFL-HCC confirmed our finding that the fusion was detectable in conventional components. An expanded screen identified a third case of fusion-positive HCC, which upon review, also had both conventional and fibrolamellar features. This screen confirmed the absence of the fusion in all conventional HCC and adjacent non-tumor liver samples. These results indicate that mFL-HCC is similar to pure FL-HCC at the genomic level and the DNAJB1:PRKACA fusion can be used as a diagnostic tool for both pure and mFL-HCC.
Team members:
Obi Griffith, Malachi Griffith, Kilannin Krysiak, Avinash Ramu, Zachary Skidmore, Jason Kunisaki
Lung cancer genomics
Recurrent WNT Pathway Alterations are Frequent in Relapsed Small Cell Lung Cancer
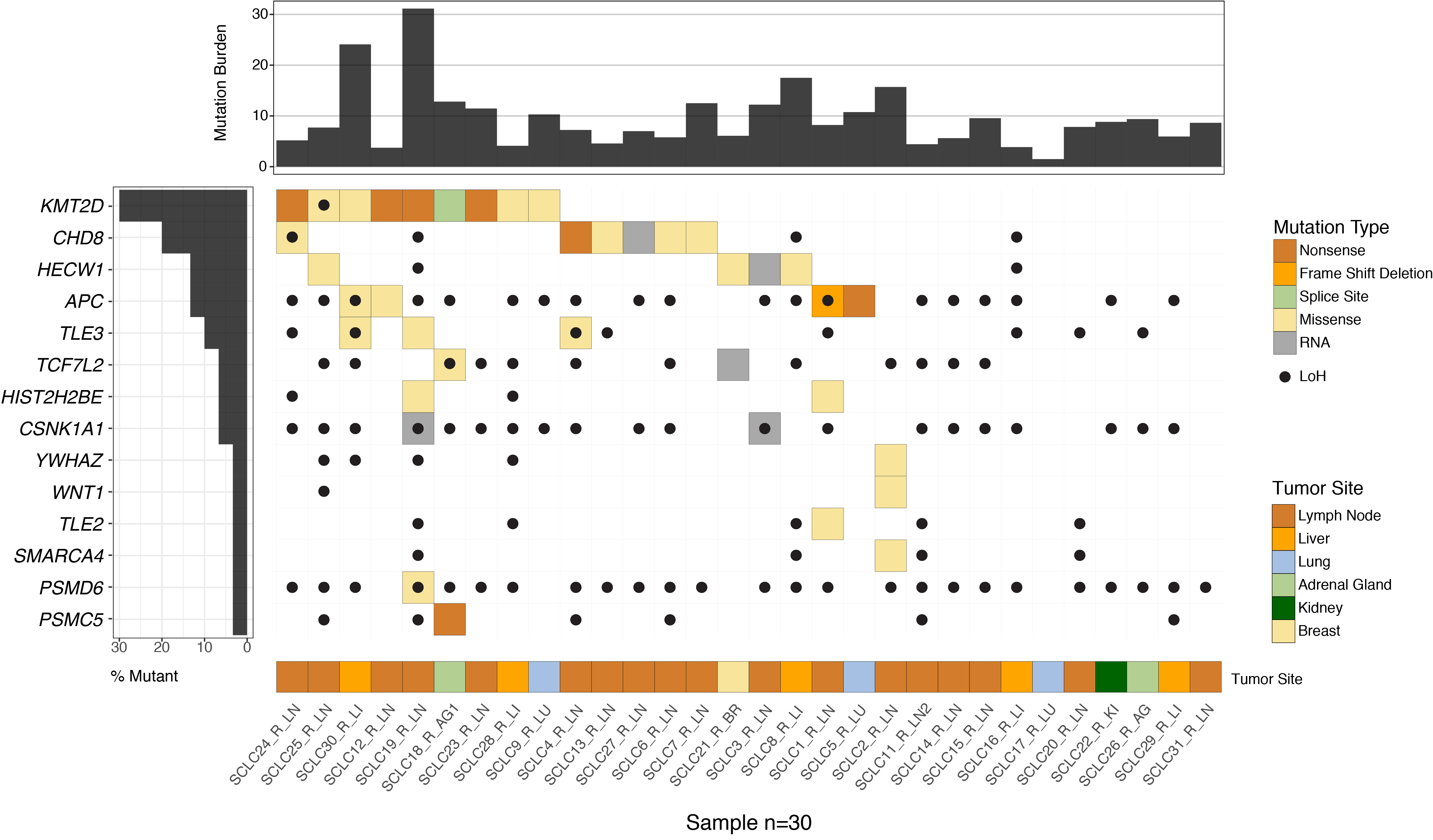
Description:
Nearly all patients with small cell lung cancer (SCLC) eventually relapse with chemotherapy-resistant disease. The molecular mechanisms driving chemotherapy resistance in SCLC remain to be characterized. We performed whole-exome sequencing of paired SCLC tumor samples procured at diagnosis (treatment-naive samples) and relapse (relapse samples) from 12 patients, along with unpaired relapse samples obtained from 18 additional patients. We observed multiple somatic copy number alterations, including gains in ABCC1, and deletions in MYCL, MSH2, and MSH6 among relapse samples. Relapse samples also showed recurrent mutations in regulators of WNT signaling, such as CHD8 and APC. Analysis of RNA-sequencing data suggested enrichment for “ASCL1-low” expression subtype and WNT activation in relapse samples. Activation of WNT signaling in chemotherapy-sensitive human SCLC cell lines through APC knockdown induced chemotherapy resistance. Additionally, in vitro-derived chemotherapy-resistant cell lines demonstrated increased WNT activity. Our results support a role for WNT signaling activation as a mechanism of chemotherapy resistance in relapsed SCLC.
Team members:
Alex Wagner, Zach Skidmore, Kilannin Krysiak, Avinash Ramu, Lee Trani, Jason Kunisaki, Nick Spies
Lymphoma genomics
Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma
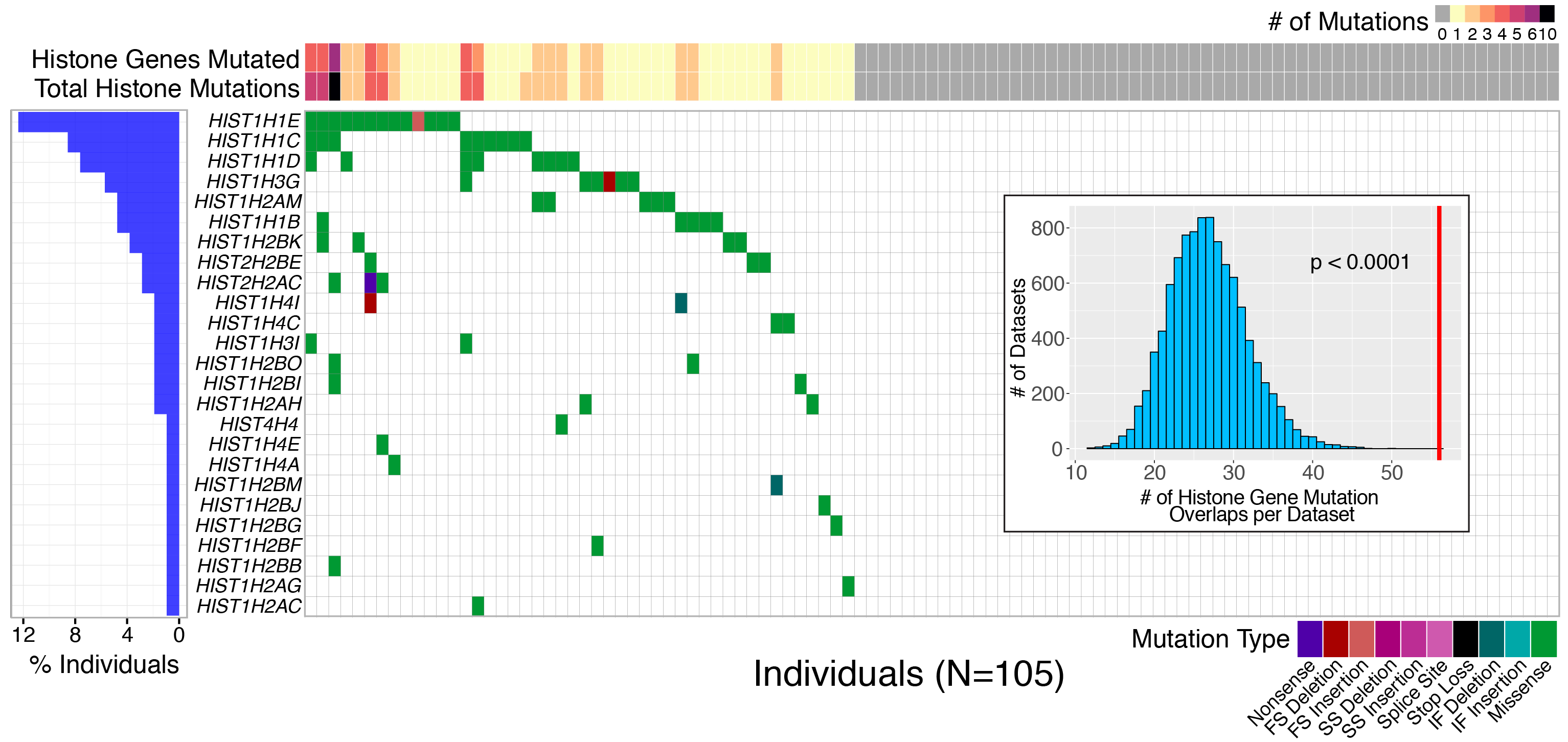
Description:
Follicular lymphoma (FL) is the most common form of indolent non-Hodgkin lymphoma, yet it remains only partially characterized at the genomic level. To improve our understanding of the genetic underpinnings of this incurable and clinically heterogeneous disease, whole-exome sequencing was performed on tumor/normal pairs from a discovery cohort of 24 patients with FL. Using these data and mutations identified in other B-cell malignancies, 1716 genes were sequenced in 113 FL tumor samples from 105 primarily treatment-naive individuals. We identified 39 genes that were mutated significantly above background mutation rates. CREBBP mutations were associated with inferior PFS. In contrast, mutations in previously unreported HVCN1, a voltage-gated proton channel-encoding gene and B-cell receptor signaling modulator, were associated with improved PFS. In total, 47 (44.8%) patients harbor mutations in the interconnected B-cell receptor (BCR) and CXCR4 signaling pathways. Histone gene mutations were more frequent than previously reported (identified in 43.8% of patients) and often co-occurred (17.1% of patients). A novel, recurrent hotspot was identified at a posttranslationally modified residue in the histone H2B family. This study expands the number of mutated genes described in several known signaling pathways and complexes involved in lymphoma pathogenesis (BCR, Notch, SWitch/sucrose nonfermentable (SWI/SNF), vacuolar ATPases) and identified novel recurrent mutations (EGR1/2, POU2AF1, BTK, ZNF608, HVCN1) that require further investigation in the context of FL biology, prognosis, and treatment.
Team members:
Kilannin Krysiak, Felicia Gomez, Matthew Matlock, Lee Trani, Malachi Griffith, Obi Griffith
Head and neck cancer genomics
Oral cavity squamous cell carcinoma xenografts
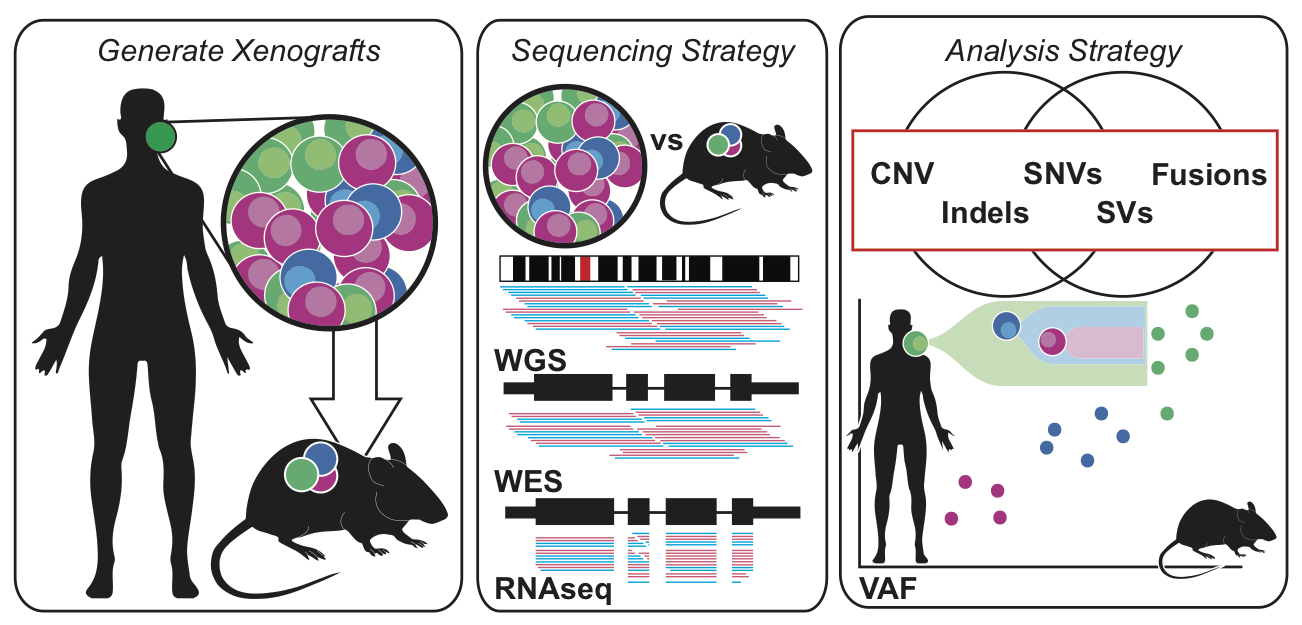
Description:
Comprehensive genomic analysis was performed on patient derived xenografts for oral squamous cell carcinomas (OSCC). We found PDX samples were largely correlative with the primary tumors from which they were derived. PDX models were able to retain the heterogeneous mutational landscape and clonal architecture of tumors. Somatic differences between the PDX and corresponding OSCC primary samples consisted primarily of low-frequency mutations, making these xenografts ideal models for exploring OSCC tumor biology.
Team members:
Katie Campbell, Zachary Skidmore, Erica Barnell, Malachi Griffith, Obi Griffith
Immunogenomics
Neoantigen characterization and personalized cancer vaccine design
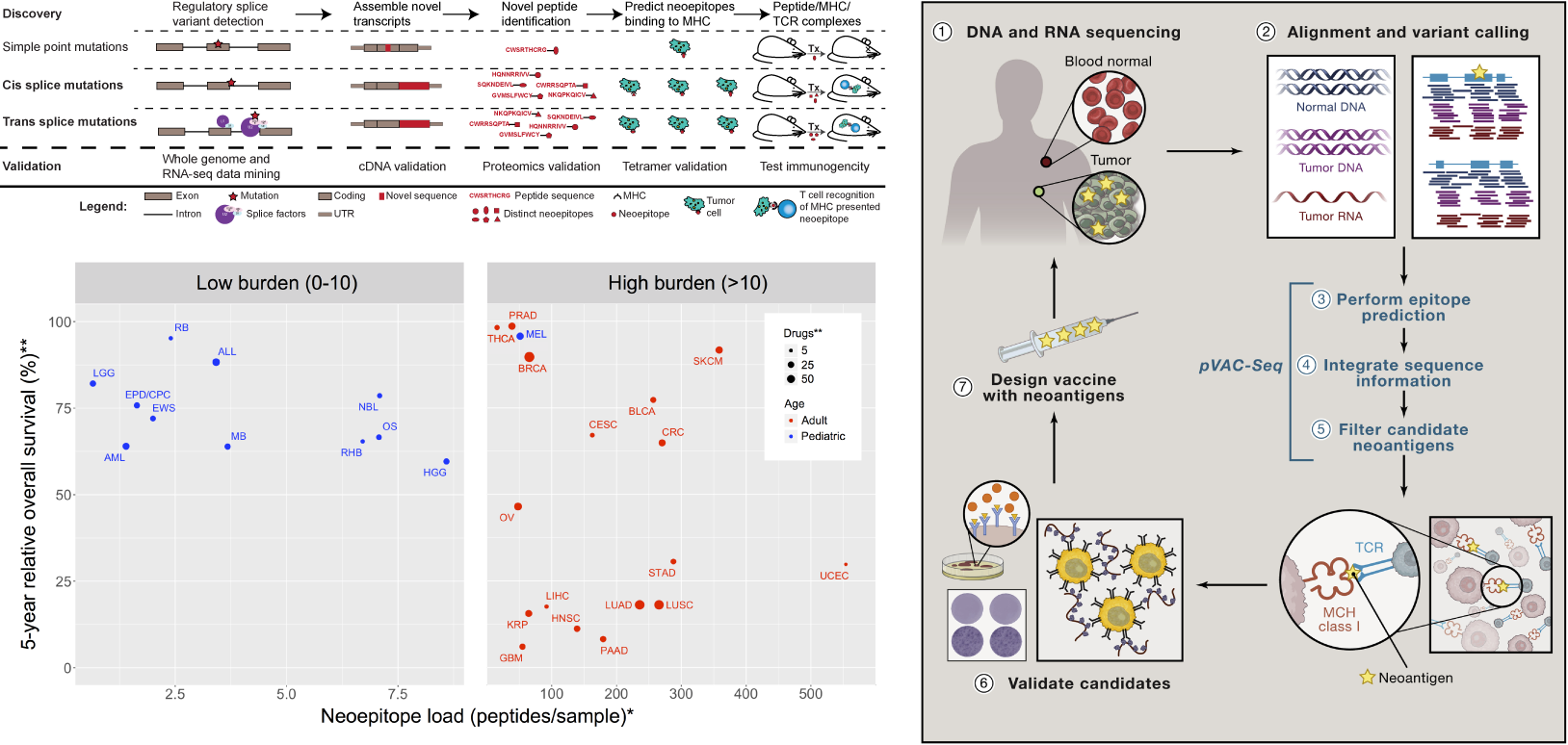
Description:
In support of several clinical trials we are creating and applying new immunogenomics tools to help predict response to checkpoint blockade inhibition therapies and design personalized cancer vaccines.
Publication(s):
Team members:
Jasreet Hundal, Katie Campbell, Yang-Yang Feng, Connor Liu, Joshua McMichael, Susanna Kiwala, Obi Griffith, Malachi Griffith
Personalized neoantigen vaccine clinical trials
As part of our interest in precision medicine and cancer genomics, we have been and are currently involved with several clinical trials at Washington University School of Medicine in which personalized cancer vaccines were designed using patient-specific neoantigens. All of the following trials are underway. All involve personalized cancer vaccines either alone or in combination with other therapies (most commonly checkpoint blockade therapy). Total estimated enrollment of all trials is: 232 patients. The majority of these are in the future, as most trials are just getting underway now.
Kidney Cancer (Renal Cell Carcinoma)
PolyImmune {Durvalumab (MEDI4736) and Tremelimumab} & Vaccine Orchestrated Treatment for Patients With Advanced/Metastatic Renal Cell Carcinoma (NCT03598816)
Lung Cancer
A Personal Cancer Vaccine (NEO-PV-01) With Pembrolizumab and Chemotherapy for Patients With Lung Cancer (NCT03380871)
Personalized Neoantigen Vaccine in Combination With Durvalumab (MEDI4736) in Extensive Stage Small Cell Lung Cancer (NCT04397003)
Brain Cancer
Neoantigen-based Personalized Vaccine Combined With Immune Checkpoint Blockade Therapy in Patients With Newly Diagnosed, Unmethylated Glioblastoma (NCT03422094)
Neoantigen-based Personalized DNA Vaccine in Patients With Newly Diagnosed, Unmethylated Glioblastoma (NCT04015700)
Neoepitope-based Personalized DNA Vaccine Approach in Pediatric Patients With Recurrent Brain Tumors (NCT03988283)
Breast Cancer
Neoantigen DNA Vaccine Alone vs. Neoantigen DNA Vaccine Plus Durvalumab in Triple Negative Breast Cancer Patients Following Standard of Care Therapy (NCT03199040)
Safety and Immunogenicity of a Personalized Polyepitope DNA Vaccine Strategy in Breast Cancer Patients With Persistent Triple-Negative Disease Following Neoadjuvant Chemotherapy (NCT02348320)
Follicular Lymphoma
Personalized Tumor Vaccine Strategy and PD-1 Blockade in Patients With Follicular Lymphoma (NCT03121677)
Prostate Cancer
Neoantigen DNA Vaccine in Combination With Nivolumab/Ipilimumab and PROSTVAC in Metastatic Hormone-Sensitive Prostate Cancer (NCT03532217)
Pancreatic Cancer
Neoantigen DNA Vaccine in Pancreatic Cancer Patients Following Surgical Resection and Adjuvant Chemotherapy (NCT03122106)
Neoantigen Peptide Vaccine Strategy in Pancreatic Cancer Patients Following Surgical Resection and Adjuvant Chemotherapy (NCT03956056)
Neoantigen Vaccines in Pancreatic Cancer in the Window Prior to Surgery (NCT05111353)
Melanoma (collaboration with UPenn/PICI)
Dendritic Cell Vaccination in Patients With Advanced Melanoma (NCT03092453)
Colorectal Cancer (collab with UPenn/PICI)
DC Vaccine in Colorectal Cancer (NCT03730948)
Targeted therapy clinical trials
Breast Cancer
A Phase I Trial of BKM120 (Buparlisib) in Combination with Fulvestrant in Postmenopausal Women with Estrogen Receptor-Positive Metastatic Breast Cancer (NCT01339442) PMID: 26563128
NeoPalAna: Neoadjuvant Palbociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor, and Anastrozole for Clinical Stage 2 or 3 Estrogen Receptor-Positive Breast Cancer (NCT01723774) PMID: 28270497
A Phase II Trial of Neoadjuvant MK-2206, an AKT Inhibitor, with Anastrozole in Clinical Stage II or III PIK3CA-Mutant ER-Positive and HER2-Negative Breast Cancer (NCT01776008) PMID: 28874413
Immunogenomic profiling and pathological response results from a clinical trial of docetaxel and carboplatin in triple-negative breast cancer (NCT02124902) & (NCT02547987) PMID: 34173924
Personalized ctDNA micro-panels can monitor and predict clinical outcomes for patients with triple-negative breast cancer (NCT02124902) PMID: 36273232
Genomic characterization of HER2-positive breast cancer and response to neoadjuvant trastuzumab and chemotherapy-results from the ACOSOG Z1041 (Alliance) trial. (NCT00513292) PMID: 28453704
A phase 1 trial of BKM120 (Buparlisib) in combination with fulvestrant in postmenopausal women with estrogen receptor positive metastatic breast cancer (NCT01339442) PMID: 26563128
Lymphoma
Phase I Trial of N-803, an IL15 Receptor Agonist, with Rituximab in Patients with Indolent Non-Hodgkin Lymphoma (NCT02384954) PMID: 33832946
Phase 1/dose expansion trial of brentuximab vedotin and lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (NCT02086604) PMID: 34780623
Single-Agent Ibrutinib in Relapsed or Refractory Follicular Lymphoma: A Phase 2 Consortium Trial (NCT01849263) PMID: 29074501
Head and Neck Cancer
Neoadjuvant and Adjuvant Pembrolizumab in Resectable Locally Advanced, Human Papillomavirus-Unrelated Head and Neck Cancer: A Multicenter, Phase 2 Trial (NCT02086604) PMID: 34780623
Variant interpretation
CIViC: a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer

Description:
CIViC is an expert-crowdsourced knowledgebase for Clinical Interpretation of Variants in Cancer describing the therapeutic, prognostic, diagnostic and predisposing relevance of inherited and somatic variants of all types. CIViC is committed to open-source code, open-access content, public application programming interfaces (APIs) and provenance of supporting evidence to allow for the transparent creation of current and accurate variant interpretations for use in cancer precision medicine.
Team members:
Malachi Griffith, Nicholas Spies, Kilannin Krysiak, Josh McMichael, Adam Coffman, Arpad Danos, Benjamin Ainscough, Cody Ramirez, Lynzey Kujan, Erica Barnell, Alex Wagner, Zachary Skidmore, Connor Liu, Rachel Bilski, Robert Lesurf, Yang Yang Feng, Lee Trani, Matt Matlock, Avinash Ramu, Katie Campbell, Greg Spies, Aaron Graubert, Jason Walker, Obi Griffith
Precision medicine
Genome analysis of relapsed adult ALL case reveals personalized therapeutic strategy
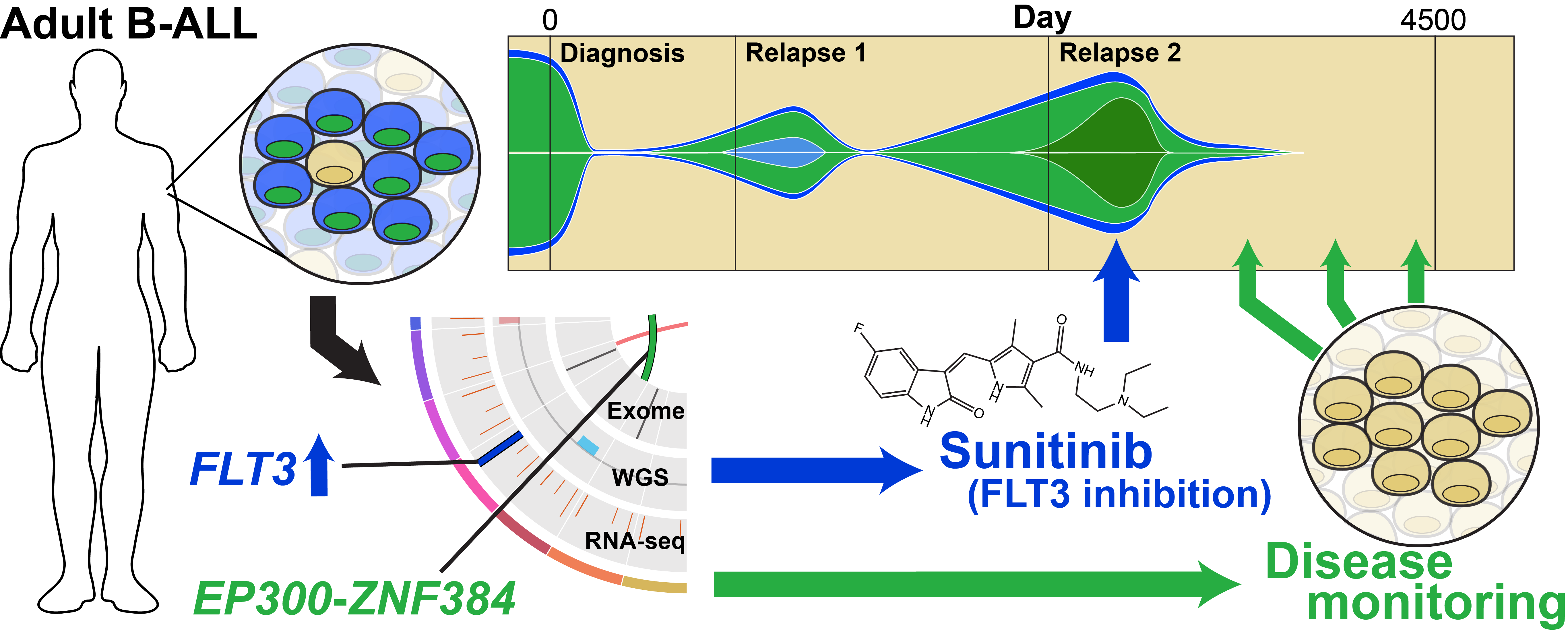
Description:
Extensive genomic analyses were performed in an adult with post-allo relapsed B-ALL. Mutations were found in EP300, NF1, IKZF1, SETD2, RB1, PAX5, NF1, ETV6, and ZNF384. Transcriptome analysis identified aberrant overexpression of the FLT3 gene. Treatment with the FLT3 inhibitor sunitinib induced a rapid clinical and molecular response. This study demonstrates a powerful proof-of-principle that comprehensive genomic studies can sometimes reveal unexpected clinically actionable therapeutic targets.
Publication(s):
Griffith et al. 2016. Experimental Hematology. 44(7):603-13.
Team members:
Malachi Griffith, Obi Griffith, Kilannin Krysiak, Zachary Skidmore, Avinash Ramu, Alex Wagner, Katie Campbell, Robert Lesurf, Jasreet Hundal, Nicholas Spies, Benjamin Ainscough, Jason Walker
Optimizing Cancer Genome Sequencing and Analysis
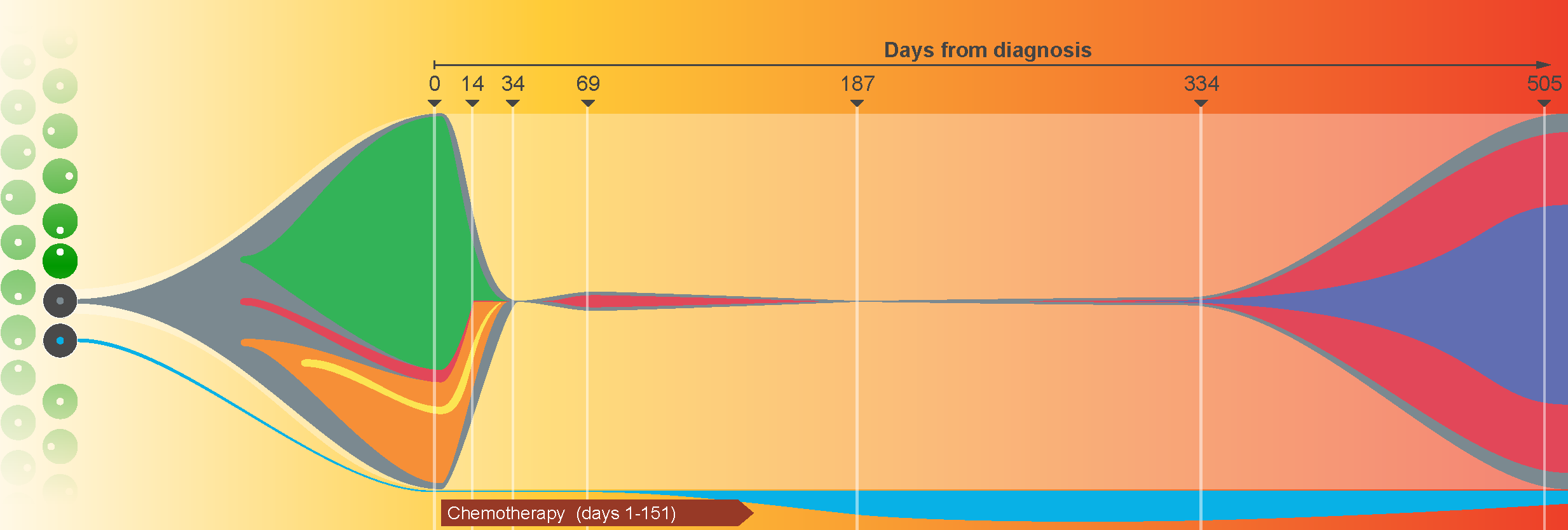
Description:
Tumors are typically sequenced to depths of 75-100x (exome) or 30-50x (whole genome). We demonstrated that current sequencing paradigms are inadequate for tumors that are impure, aneuploid or clonally heterogeneous. To reassess optimal sequencing strategies, we performed ultra-deep (up to ~312x) whole genome sequencing (WGS) and exome capture (up to ~433x) of a primary acute myeloid leukemia, its subsequent relapse, and a matched normal skin sample. We tested multiple alignment and variant calling algorithms and validated ~200,000 putative SNVs by sequencing them to depths of ~1,000x. Additional targeted sequencing provided over 10,000× coverage and ddPCR assays provided up to ~250,000x sampling of selected sites. We evaluated the effects of different library generation approaches, depth of sequencing, and analysis strategies on the ability to effectively characterize a complex tumor. This dataset, representing the most comprehensively sequenced tumor described to date, serves as an invaluable community resource.
Publication(s):
Team members:
Malachi Griffith, Obi Griffith, Kilannin Krysiak, Zach Skidmore, Avinash Ramu, Jason Walker, Lee Trani